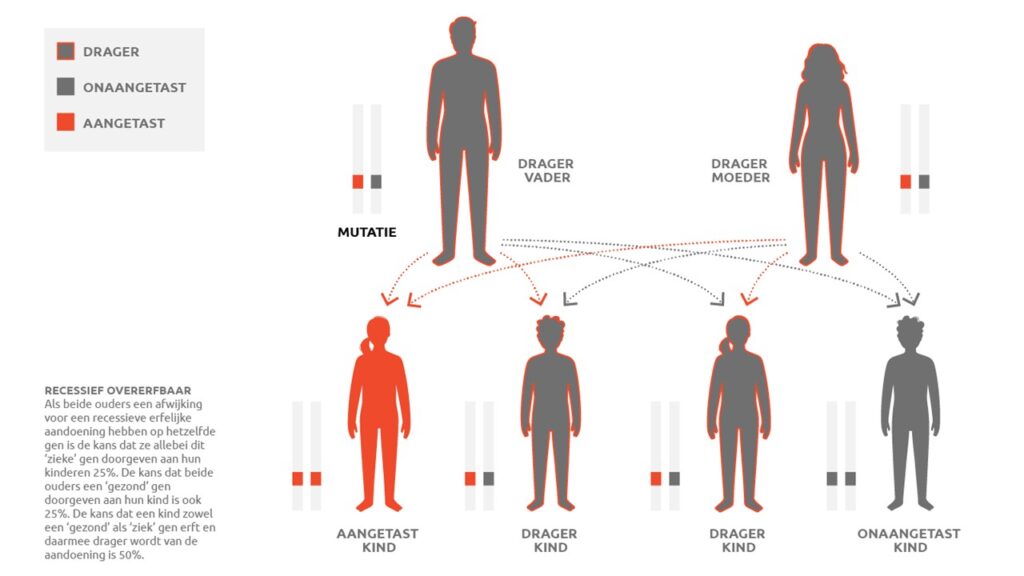
De oorzaak voor het niet of niet goed werken van deze enzymen, die verantwoordelijk zijn voor de afbraak van afvalstoffen, is bijna altijd een genetisch defect. Bijna alle lysosomale stapelingsziekten zijn autosomaal recessief overerfbaar. Recessief overerfbaar betekent dat zowel de vader als de moeder drager zijn van de ziekte voordat de ziekte tot uiting kan komen bij hun kind. Een drager heeft zelf geen ziekteverschijnselen. Autosomaal betekent dat jongens en meisjes een even grote kans hebben op het krijgen van de ziekte. Een kind heeft een kans van 25 procent op het krijgen van de ziekte. In 50 procent van de gevallen wordt het kind drager en de overige 25 procent is gezond.








Plaats een opmerking